What is 3D-BLAST
|
3D-BLAST is a very fast and accurate method for discovering the homologous proteins
and evolutionary classifications of a newly determined protein structure. Our 3D-BLAST
has the advantages of BLAST tool for fast protein structure database scanning.
It searches for the longest common substructures, called SAHSPs (structural alphabet
high-scoring segment pairs), existing between the query structure and every structure
in the structural database. The SAHSP is similar to the high-scoring segment
pair (HSP) in BLAST. The 3D-BLAST ranks the search homology structures based
on both SAHSP and E-value calculating from the substitution scoring matrix of
structural alphabets. With regard to sensitivity and selectivity of the structural
matches, 3D-BLAST compares well to the related programs, although it is by far faster.
Our method search more than 10000 protein structures in 1.3 seconds and achieved a good agreement with the results of detailed structure alignment methods.
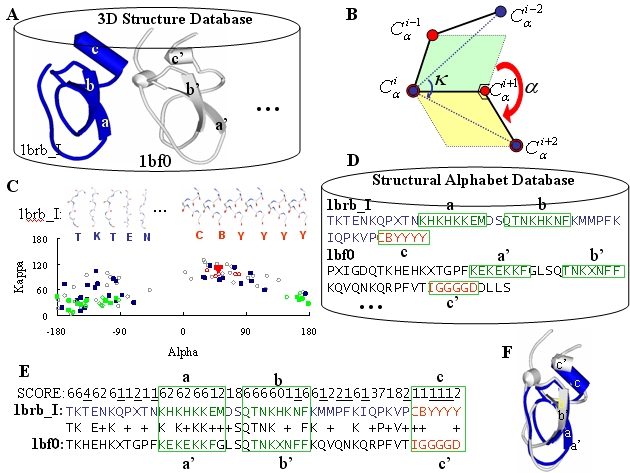
The following Figure shows the outline of 3D-BLAST for fast scanning a library of a
structural alphabet sequence database (SADB), which is coded from known protein
structures. Here, we used two proteins, 1brb with I chain (1brb_I, blue) and 1bf0
(gray), to describe these steps and concepts. First, we divided a 3D protein
structure into 3D protein fragments, each five residues long called a structural
alphabet, by using kappa
(k)
and alpha
(a)
angle (Figure B) defined as in the DSSP program. According to the
k and
a angles, each structure in the protein
structure database has a specific
(k, a)-map
distribution (Figure C) and is able to be encoded into a corresponding 1D
structural alphabet sequence collected in the SADB database (Figure D).
Third, we used a generalized theory of a substitution
matrix to develop a new structural alphabets substitution matrix
(SASM). We then enhanced the sequence alignment tool, BLAST, which searches on
the SADB by using the SASM to fast discover the protein structure homology or evolutionary classifications.
The resulting structural alphabet sequence alignment (Figure E) was reported with
E-value as the BLAST, and the structure alignment (Figure F) was also yielded.
Figure C shows that the
(k, a)-map
distributions of 1brb_I (filled squares) and
1bf0 (empty circle) are similar. The strand structures (green) and helix structure
(red) of these two proteins are aligned by the 3D-BLAST and their aligned structures
are also similar even though their sequence identity is 21.3%.
|
|
|
|
|
|
Step-by-step illustration of the 3D-BLAST using the protein 1brb chain I as the query
protein searching against nrPDB. (A) A known three-dimensional database with two
structures, 1brbI (blue) and 1bf0 (gray). (B) The definitions of the kappa
(k) and alpha
(a) angles.
The k
, ranging from 0° to 180°, of a residue i is a bond angle formed by three
Ca
atoms of residues i-2, i, and i+2.
The a
, ranging from -180° to 180°, of a residue i is a dihedral angle formed by the four
Ca
atoms of residues i-1, i, i+1, and i+2. (C) The
(k, a)-maps
of 1brbI (square) and 1bf0 (circle) are the similar. The strand (green)
and the helix (red) are indicated. The 3D-structure fragments of the first five
and last five of 1brbI are given. (D) The structural alphabet sequence database
(SADB). (E) The result and score of aligning two structural alphabet sequences
using BLAST and the structural alphabet substitution matrix (see text). For example,
the scores of aligning T to T is 6, K to K is 6, and T to H is -4. (F) The
resulting structure alignments of the solution identified in (E).
|
|
